Multi-perspective Genomics Research: Scientists use COVID-19 Host Genetics Initiative genomic data to characterize COVID-19
July 07, 2021
Written by Gita Pathak, PhD; Annika Faucon B.S.; Atanu Kumar Dutta, MD
Edited by Kumar Veerapen, PhD and Brooke Wolford, PhD
The Coronavirus Disease 2019 (COVID-19), caused by novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has resulted in a pandemic claiming the lives of more than 3.2 million individuals and infecting more than 156 million individuals worldwide (WHO: https://covid19.who.int/). A complete timeline is reviewed in Figure 1 (Wiersinga et al. 2020). The SARS-CoV-2 infection has variable severities in people, ranging from asymptomatic to severe life-threatening respiratory failure, coagulation, or neurological symptoms among other sequelae. This individual level variability in response to the infection has led scientists to investigate effects of genetic variation in the human genome that may explain the wide-spectrum of COVID-19 severity and susceptibility The COVID-19 Host Genetics Initiative (HGI) has brought together scientists and studies from all over the world to identify the genomic regions associated with COVID-19 symptoms. This large-scale effort periodically collects descriptive statistics of genotype associations with three sets of COVID-19 severity thresholds:
- very severe respiratory COVID-19 confirmed (A1/A2)
- hospitalization due to COVID-19-related symptoms (B1/B2)
- individuals with laboratory confirmation of SARS-CoV-2 infection (C1/C2)
(see phenotype definitions here)
Results from these severity phenotype definitions from multiple studies across the world (see contributing studies) are analyzed together (i.e., meta-analyzd). The meta-analysis of each phenotypic definition is publicly available to everyone who would like to pursue scientific inquiries.
While many investigations have been launched using the genetic study results released by the HGI, here we collected and summarized a selection of the studies (last searched on June 14, 2021). The goal of this post is to introduce the extent of scientific studies made possible through the open-access and collaborative initiative of COVID-19 HGI.
Disclaimer: Some of these studies are still posted as preprints and have not been peer-reviewed, therefore should not be used to guide clinical or therapeutic practice. The writing in this post does not reflect the opinion of the authors whose study is discussed. Please read the original links for each study.
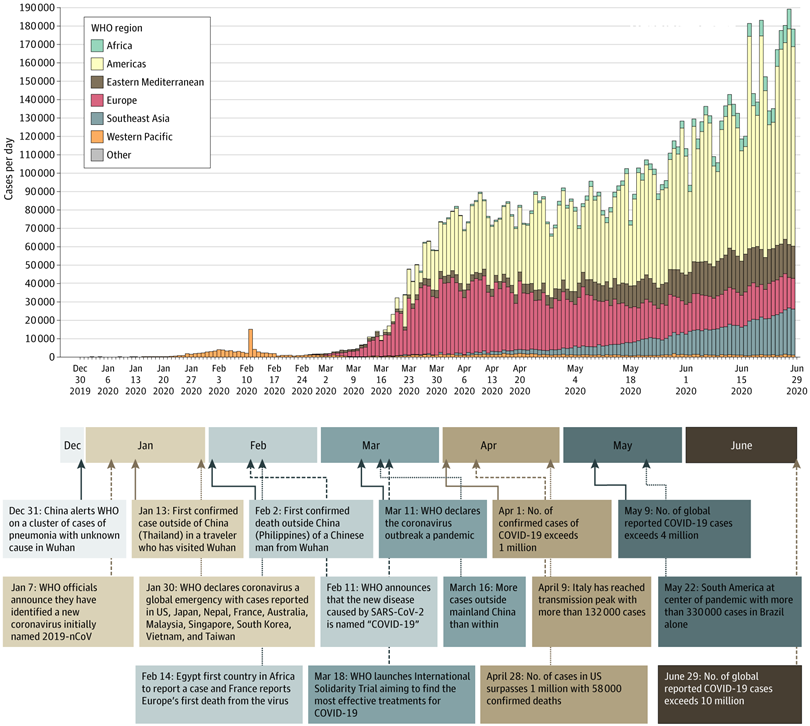
Figure 1: Overview of COVID-19 timeline, please see (Wiersinga et al. 2020) for full text.
Multi-omics investigations of COVID-19
Multi-omics investigations involve the integration of multiple layers of omics data (e.g. genomic, transcriptomic, proteomic). For example, genotypic variation is associated with gene splicing, and/or protein expression of genes within cis (local: 1MB) regions. In the following studies authors combined results of functional studies to make statistical predictions to learn about the pathology of COVID-19.
Yunlong Ma and colleagues used the results of Freeze 2 data from HGI together with genotype summary-level data from another study (Ellinghaus et al) to identify a locus- 21q22.11. This locus has a variant (rs9976829) that affects splicing and is located within IFNAR2 and upstream to IL10RB. Cell-type composition analyses for the IFNAR2 expression showed an enrichment of dendritic cells, and for the IL10RB expression, an enrichment of nonclassical monocytes.
Combining results from multiple studies can increase confidence about the biological importance of a genetic feature and the need for further inquiry about how that information can be used to improve care.
Another study by Gita Pathak and colleagues extended the multi-omics investigation using gene, splicing, and protein expression data with the genotype summary statistics from Freeze 4 release of HGI. The authors also systematically tested the phenome-wide associations of genetically regulated expression in Vanderbilt Biobank, observing coagulation-related clinical symptoms, immunologic, and blood-cell-related biomarkers which were tested in pan-ancestry biobanks such as UK Biobank and Biobank Japan. These consistent associations can highlight genes and pathways involved in disease pathophysiology and give insight into disease mechanisms, and treatment.
A single-cell transcriptomic investigation by John Fullard and colleagues analyzed brain tissues (dorsolateral prefrontal cortex, medulla oblongata and choroid plexus) of COVID-19 affected patients. The microglial activation was compared to genetically regulated expression profiles of brain and blood tissues using HGI summary statistics. This identified an over-representation of microglia-specific transcription factors, suggesting neuroinflammation in the brain tissues of COVID-19 patients.
Matteo D’Antonio and colleagues investigated all loci associated with COVID-19 outcomes and tissue specific gene expression for 48 tissues to dissect the molecular mechanisms underlying potential genetic predisposition to SARS-Cov-2.

Immunological links to COVID-19
The severe immune response mediated in response to COVID-19 has led researchers to investigate genetic links between inflammatory biomarkers and COVID-19. Susanna Larson and colleagues investigated seven single nucleotide polymorphisms in and around IL6R locus and reported its association with lower C-Reactive Protein, Fibrinogen, IL-6 and soluble IL-6 receptor levels. They used COVID-19 HGI meta-analysis summary statistics (release 4) and the corresponding phenotypic data on pneumonia from the FinnGen consortium and the UK Biobank cohort for the study’s Mendelian Randomization analysis (Read more about how Mendelian Randomization works here). Their results showed that genetically proxied IL-6R inhibition reduced the risk of very severe COVID-19 (odds ratio 0.94 [95% CI - 0.89-1.00]) but increased the risk of pneumonia (odds ratio 1.04 [95% CI - 0.99-1.08]). The authors therefore cautioned against the use of IL-6 receptor blockers for the treatment of severe COVID-19.
Yitang Sun and colleagues used a two-sample bidirectional Mendelian Randomization (MR) approach to investigate causal associations between white blood cell traits and COVID-19 susceptibility and severity. They used the GWAS summary statistics of COVID-19 HGI release 5 and summary statistics for WBC traits from NHGRI-EBI Catalog of human genome-wide association studies. 115 SNPs for basophil percentage and 469 SNPs for monocyte count were selected as genetic instruments. Using the groups of very severe respiratory and hospitalized COVID-19, the study demonstrated that lower basophil count, basophil percentage, myeloid WBC count, and total WBC count increased the risk of severe COVID-19. However, none of the white blood cell traits were causally associated with COVID-19 susceptibility. Overall, this work provided insights that individuals with a lower genetic capacity for basophil production are likely at risk of the severe forms of COVID-19, and that enhancing the production of basophils may be a potential prevention and treatment strategy.
Yao Zhou and colleagues sought to infer the causality from 12 specific coagulation factors to COVID-19 severity and the underlying mechanism. These authors used Mendelian Randomization methods to better understand which statistical associations might have a causal link. Their results suggest that the associations between coagulation factors VWF/ADAMTS13 and COVID-19 severity are causal. The authors also validated their results in an independent cohort from UK Biobank.
Benjamin Schmiedel and colleagues, retrieved genetic variants (lead SNPs) associated with COVID-19 from COVID-19 HGI data release 4 (p-value < 5 x 10-8) and identified SNPs in tight linkage with the lead SNPs in the 1000 Genome project data for different ethnicities. These SNPs were then analyzed for overlap with eQTLS in the DICE database (http://dice-database.org) and 3D cis-interactome maps. They identified COVID-19 risk variants associated with expression of 11 protein coding genes including that of CCR2 (in the 3p21.31 risk locus) in classical monocytes. This is likely mediated through genetic factors that are very close to the gene on the genome and potentially only used in monocytes. Their findings point to a potentially important role for IL-10 signaling and NK cells in influencing susceptibility to severe COVID-19 illness.
Seyedeh M. Zekavat and colleagues identified a possible role of age-related mosaic chromosomal alterations to be associated with COVID-19 hospitalization.
Druggable genome and COVID-19
A common approach to identifying medications for a disease is to find out what biological differences there are between people with and without the disease and then to try to give a drug that heads off that change. Drug repurposing studies compare knowledge about the biological changes a drug imposes on the body and how those changes relate to the difference between sick and healthy individuals. Liam Gaziano and colleagues compared the list of all genes whose expression can be affected with medications, and compared the list with those associated with risk of COVID-19 hospitalization. The goal with this analysis is to highlight gene targets (and subsequently, existing medications) that can be repurposed for use in patients with COVID-19.
By looking at the data this way, the authors identified three genes whose expression might affect COVID-19 risk: ACE2, IL10RB, and IFNAR2. ACE2 codes for the ACE-2 receptor used by the virus to enter cells. IL10RB and IFNAR2 are both related to immune function, but because their genes are so close to each other and GWAS signals are shared, further analysis is needed to determine which gene was more likely to be directly impacting risk of COVID-19 hospitalization.
CM Schooling and colleagues used HGI results to determine the potential for success for three putative therapies for COVID-19: tocilizumab, statins, and anakinra. The authors used a single genetic variant with a similar effect as each of the drugs as a proxy for that drug. For instance, statins lower low density lipoprotein cholesterol, so they used a genetic variant that had a strong effect in lowering LDL cholesterol. Though this type of approach is validated in the literature, the authors also demonstrated this approach with dexamethasone, which had been demonstrated to be effective against severe COVID-19. The authors then checked whether the proxy for each drug was predictive and was associated with decreased risk for COVID-19, COVID-19 hospitalizations, or severe COVID-19. Ultimately, the authors were able to use this approach to encourage development of tocilizumab, statins, and dexamethasone—all of which have mounting evidence of effectiveness in treating COVID-19.
One of the key insights in the mode of transmission for SARS-Cov is the ACE-2 receptor protein. The structural dimerization of ACE-2 with spike proteins of SARS-CoV-2 (Yan et al. 2020) presented an important avenue for further investigation. Gita Pathak and colleagues examined the ACE-2-interacting gene network to determine whether genes that interact with ACE2 are statistically over-represented among associations with certain phenotypes, drugs, and miRNA expression. The drug-gene interaction analysis identified drugs such as dexamethasone, spironolactone, metformin and melatonin. The pathway analysis of the identified drugs showed platelet sensitization by low-density lipoprotein cholesterol, interleukin-7 cytokine and viral RNA synthesis.
Shared mechanisms between COVID-19 and other diseases
Individuals with prior medical conditions and older adults have increased risk of developing severe illness and/or longer recovery from COVID-19 infection (CDC, 2019) . The following investigations focused on genetic mechanisms for traits with disease surveillance links to COVID-19.
Guillaume Butler-Laporte and colleagues investigated the effect of 25-hydroxy Vitamin D3 deficiency with COVID-19 severity in a two sample Mendelian Randomization model. They identified genetic variants explaining serum Vitamin D levels from their previous GWAS study and then went on to test these instruments in the COVID-19 HGI statistics for a total of 81 variants. They found genetically increased 25-hydroxy Vitamin D3 levels did not protect against COVID-19 susceptibility, hospitalization, or severity.
Alexander Hatoum and colleagues wanted to test if genetic liability to Cannabis Use Disorder (CUD) correlates with risk of COVID-19 hospitalization. To this end, they used GWAS summary statistics of CUD and COVID-19 HGI to show that these two traits are correlated and share a common genetic factor using genomic structural equation modelling.
João Fadista and colleagues determined the genetic correlation between idiopathic pulmonary fibrosis (IPF) and severe COVID-19. They performed a two-sample Mendelian Randomization study involving instruments from a previous IPF GWAS study and COVID-19 HGI summary statistics. They found that the genetic variant presenting the greatest risk for IPF (MUC5B variant rs35705950) had a protective effect against COVID-19 severity. All other instruments combined had a causal effect for COVID-19 severity. The authors therefore postulate that antifibrotic therapies used to treat IPF could have an important role in mitigating COVID-19 severity in IPF patients.
Frank Wendt and colleagues calculated the genetic correlation between three COVID-19 severity phenotypes and the set of 7,218 phenotypes in the UK Biobank with at least 50 cases. The authors used genetic correlation and latent causal variable analysis to identify putative shared links and causal relationships between COVID-19 susceptibility and several traits. The authors report evidence of causal relationships for presence of depressive symptoms, metformin use, and alcohol use as top correlates with COVID-19 severity phenotypes. A phenome-wide investigation of COVID-19 associated loci identified several laboratory measures such as alkaline phosphatase, low density lipoprotein cholesterol,hemoglobin concentration, and lymphocyte count.
Several studies have reported the role of obesity with COVID-19 severity outcomes. Brenda Cabrera Mendoza and colleagues performed genomic causal analysis using Mendelian Randomization approach to identify the role of socioeconomic status on obesity-associated traits. Using the data on self-reported household income from the UK Biobank and the COVID-19 genetic data from the HGI, the authors report that the effect of BMI and waist circumference on COVID-19 outcomes is not independent of socioeconomic status.
Another study investigating the metabolic effects on COVID-19 was performed by Noah Lorincz-Comi & Xiaofeng Zhu, who reported potential genetic causal association of type-2 diabetes and pulse pressure with COVID-19 hospitalization using Mendelian Randomization approach.
Tomoko Nakanishi and colleagues characterized the age-dependent effect of genetic variants associated with COVID-19 mortality and susceptibility. The major locus on chromosome 3 that is associated with COVID-19 severity is more evident in individuals under 60 years of age.
An innovative study design by Irene V. van Blokland and colleagues used the HGI summary statistics to replicate the genetic associations of predicted COVID-19 symptoms and severity.
Evolutionary perspective of COVID-19 genetics
Because of backcrossed mating and introgression thousands of years ago, genetic variants that can be traced to ancient hominids, such as Neanderthals and Denisovians, exist in modern day humans. With the help of scientific advancements that allow the study of ancient DNA, we can compare the genetic profile of our ancestors such as Neanderthals with modern day humans to identify which parts (haploblocks) of our genome are similar and therefore likely ancestrally-derived. With each generation, genomic chunks are broken down into smaller segments, called haploblocks, and the rate at which these haploblocks are formed is identified as recombination rate. These measurements allow us to statistically evaluate the probability of a certain haploblock to have ancestral origins. Hugo Zeberg and Svante Pääbo investigated the haploblock on the chromosome 3 COVID-19 associated locus for its possible Neanderthal origins. The authors identified a 50 kilobase region on the chromosome 3p21 to have Neanderthal origins in the individuals with European and South Asian genetic ancestry. The authors identified another region of 75 kilobases on chromosome 12 which had Neanderthal origins, and increased prevalence in Eurasia.