Data Freeze 6 Summary
July 30, 2021
COVID-19 HGI Results for Data Freeze 6 (June 2021)
Written by Chloe Kirk, BS and Guillaume Butler Laporte, MD
Edited by Brooke Wolford, PhD and Kumar Veerapen, PhD
Note: The COVID-19 Host Genetics Initiative (HGI) represents a consortium of over 3000 scientists from over 54 countries working collaboratively to share data, ideas, recruit patients and disseminate our findings. For a primer on our study design, please read our inaugural blog post. Our research is iterative, and we summarize our new results via blog posts and on the results section of our website. Finally, if any vocabulary here is unfamiliar, please send us an email at hgi-faq@icda.bio—we’d be happy to update the information here to provide more clarity. You can also check out our FAQ and take a look at this resource to review the basics of genetics.
The COVID-19 HGI Study Overview
The COVID-19 Host Genetics Initiative (HGI) is an international team of scientists focused on elucidating human genetic variations that influence responses to SARS-CoV-2 infection and its disease, COVID-19. The study is based on accumulating DNA samples from COVID-19 patients all across the world and comparing them to negative controls. This type of study is referred to as a Genome Wide Association Study (GWAS).You can find more information about how this study was conducted and past data freeze results in the previous blog posts: Data Freeze 3, Data Freeze 4, Data Freeze 5 (what is a data freeze? Look here). Our flagship manuscript based on data freeze 5 was recently published in Nature and you can read more about the paper here.
Latest Data Freeze 6 (June 2021) Increases Sample Size and Genetic Robustness in Study
As in previous data freezes, the more samples the COVID-19 HGI accumulates, the more robust we consider the GWAS findings to be. We have doubled the sample size since the last data freeze in January 2021, with a total of 25,027 COVID-19 hospitalized patient samples and 2,836,272 control samples. Additionally, this is the most diverse data freeze yet including new studies from Egypt, Saudi, Iran, India, and Thailand resulting in a total of 61 studies from 24 countries. This meta-analysis study represents one of the largest GWAS in history with greater than 2 million individual samples and over 3,300 scientists participating.
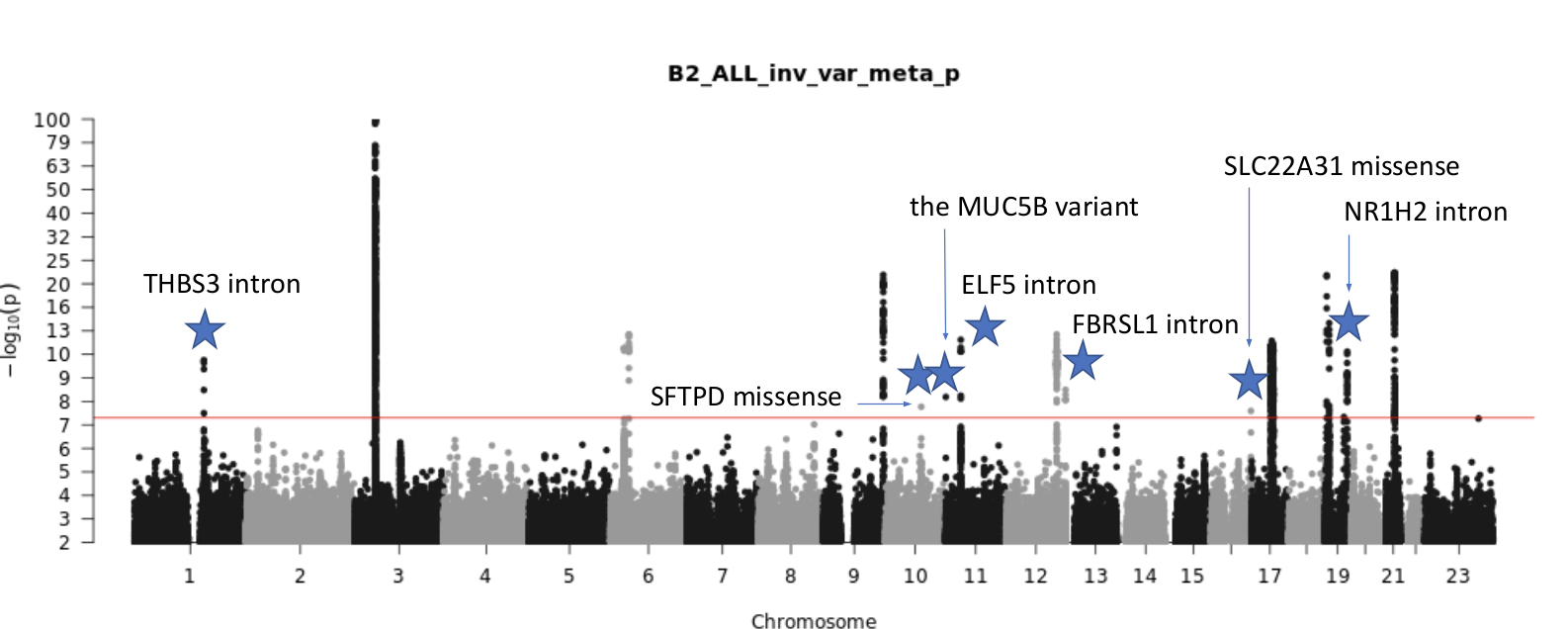
Figure 1: Current results from Data freeze 6 (June 2021). In this Manhattan plot, the points represent p-values for genome wide genetic variants, calculated by comparing the frequency between 25,027 cases (patients who were hospitalized from COVID-19) and 2,836,272 controls (samples from the population presumed to be COVID-19 negative).
New Findings Indicate Lung Genetic Mutations as Possible Contributors to Developing Severe COVID-19
Seven new genetic positions (a.k.a. variants) are significantly associated with COVID-19 hospitalization in this data freeze on chromosomes 1, 10, 11, 12, 16, and 19 (noted by blue stars in Figure 1). Some of these are associated with decreased lung function or surfactant proteins, which contribute to immune system function in the lungs. Find more details on these new and exciting findings below:
Intronic Variant in THBS3
The most statistically significant variant on chromosome 1 is an intronic variant in Thrombospondin 3 (THBS3). Using data from the Gene-Tissue Expression Project (GTEx), this variant is associated with increased THBS3 expression in lung tissue, decreased GBAP1 expression in lung tissue, and decreased MUC1 expression in other tissues. Variants in this region are known to decrease lung function (NCBI THBS3, Schips et al., 2015), and there is also a missense variant in the MTX1 gene that is highly correlated with the intronic variant.
Missense Variant in SFTPD
The gene for Surfactant Protein D, SFTPD, on chromosome 10 has a significant missense variant. This gene is associated with decreased lung function and previously associated with chronic obstructive pulmonary disease, COPD (NCBI SFTPD, Obeidat at al., 2017). This protein confers protection from severe COVID-19 symptoms, as seen in a study using SFTPD to see inhibition of interactions with SARS-CoV-2 spike protein (Fadista et al., 2021).
Upstream Variant in MUC5B
Mucin 5B, MUC5B, on chromosome 11 is known to have risk variants for idiopathic pulmonary fibrosis (NCBI MUC5B, Yang et al., 2015). The protein MUC5B is required for airway defense by encoding for mucin glycoproteins (Roy et al., 2015).
Intronic Variant in ELF5
The intronic variant found in ELF5 is also on chromosome 11 (as in MUC5B above). This protein regulates epithelium specific genes and is a member of the epithelium-specific subclass of the ETS transcription factor family (NCBI ELF5). It has no direct lung trait associations, but the gene has been previously associated with poor SARS-CoV-2 prognosis (Chen and Zhong, 2020).
Intronic Variant in FBRSL1
Fibrosin like 1, FBRSL1, on chromosome 12 has no known lung trait associations and is a poorly characterized gene (NCBI FBRSL1). This finding is quite exciting because of how little is understood of this gene so far, and this discovery provides a new clue for FBRSL1’s function.
Missense Variant in SLC22A31
Solute carrier family 22 member 31, SLC22A31, chromosome 16 has a significant missense variant. SLC22A31 is co-expressed with surfactant proteins, which are a critical risk factor for COVID-19 when surfactants are lowered or altered (Zhong et al., 2020, Weiskirchen, 2020).
Genetic variation in ACE2 associated with COVID-19
In our GWAS meta-analysis of all COVID-19 cases (125,584 cases versus 2,575,347 controls) we identified significant variants near ACE2. This is exciting because of ACE2’s role in SARS-CoV-2 infection—first discussed in data freeze 3 (read link to learn more about ACE2) from July 2020 with its relationship to a variant discovered in SLC6A20. In short, ACE2 is a receptor for SARS-CoV-2 binding, and variants on this receptor would affect how tightly SARS-CoV-2 binds to our cells (R&DSystems, ACE-2 receptor for SARS-CoV-2).
The most significant variant is rare in contributing studies of European ancestries, but more common in studies of South Asian ancestries—another great example of the importance of global representation in this analysis. While this rare variant is seven orders of magnitude more significant than other variants in the region, there are independently significant common variants nearby in the intron of CA5PB1.
Rare variants in TLR7 likely involved in severe COVID-19
Complementary to the COVID-19 HGI’s work on common genetic variants, the whole-exome/whole-genome working group uses next-generation sequencing technology to find rare genetic variants (present in less than 0.1% of the population) that may lead to worse COVID-19 presentations. Studying rare variants requires large sample sizes, and the working group led by Brent Richards (McGill) and Alessandra Renieri (Sienna) has assembled the world’s largest cohort of patients with exome sequencing, tallying more than 3,400 severe cases and 500,000 controls. Preliminary results show a likely role of TLR7 in severe COVID-19, an immunity related gene involved in the detection of viral RNA. Patients with rare deleterious variants in that gene may have an increased odds of severe disease that is 17 times higher than those without such variants. However, more replication is needed, and the third data freeze is expected to be released in August of 2021.
Our Next Steps
This sixth data freeze provides the largest insight yet into COVID-19 host genetics. These exciting new discoveries highlight the importance of international collaboration—the COVID-19 HGI includes 54 contributing studies including over 3,000 scientists worldwide. As always, our goal is to continue broadening the dataset with more samples to not only replicate these findings, but to identify other variants contributing to COVID-19 susceptibility and severity. Potential long term applications of these genetic findings could lead to improved COVID-19 treatments, prediction of individuals most at risk for severe COVID-19, and management of COVID-19 patients.
Acknowledgements
Thank you to Andrea Ganna, PhD for thoughtful feedback and comments.