This is a translation in French. You can also read the original English version.
French-3rd-blog
April 07, 2021
Résultats de la COVID-19 HGI pour le gel de données 5 (Janvier 2021)
2 mars 2021
Rédigé en anglais par Minttu Marttila, Annika Faucon, Nirmal Vadgama, Shea Andrews, Brooke Wolford, and Kumar Veerapen traduit en français par Palwendé Romuald Boua, Marie-Julie Favé, Audrey Lemaçon, Sophie Limou, et Isabelle Migeotte au nom de la COVID-19 HGI
Note: La COVID-19 Host Genetics Initiative (HGI) est un consortium formé de plus de 2000 chercheurs de plus de 54 pays collaborant pour partager données et idées, recruter des patients et diffuser les résultats de leurs analyses. Pour un aperçu de la conception de notre étude, vous pouvez vous référer au premier article de notre blog (inaugural blog post). Nos résultats évoluent rapidement, et nous les résumons périodiquement sur notre blog (blog posts) et sur la section des résultats (results) de notre site web. De plus, le but de cet article étant de vulgariser nos efforts, n’hésitez pas à nous contacter si vous avez des questions sur son contenu ou le vocabulaire utilisé et nous serons heureux d’y apporter toute clarification nécessaire (hgi-faq@icda.bio). Lors des prochaines semaines, nous ajouterons aussi des informations additionnelles expliquant certains concepts ou terminologies utilisés par la COVID-19 HGI. En attendant, nous vous invitons à visiter la ressource suivante pour une revue des principes de base de la génétique (en anglais, et en français).
La publication scientifique présentant les résultats de ce gel de données est maintenant disponible sur medRXiv.
Le dernier gel de données (version 5) augmente la taille de l'échantillon et la robustesse des découvertes génétiques
La COVID-19 HGI a découvert de manière itérative des signaux génétiques robustes dans nos versions précédentes et représente la plus grande étude d'association pangénomique (GWAS, ressource en anglais et en français) de l'histoire: tant en termes de participants à l'étude (> 2 millions d'individus) que de nombre de collaborateurs (> 2000 scientifiques). Ici, nous décrivons les résultats issus de notre dernier gel de données (version 5). Dans notre précédent gel de données (version 4), nous avons rapporté l'identification de variants génétiques humains associés à une COVID-19 sévère (voir nos articles de blog vulgarisés présentant les résultats pour les versions 3 et 4). Nous avions identifié ces variants grâce à un GWAS chez plus de 30000 patients atteint de la COVID-19 (les cas) et 1,47 million de patients non atteints de la COVID-19 (les contrôles). Dans le gel de données version 5, nous avons encore augmenté la taille de l'échantillon à près de 50000 cas de COVID-19 et à plus de 2 millions de contrôles en combinant les données de 47 études menées à travers 19 pays (Figure 1). En augmentant la taille de l'échantillon, nous améliorons également la fiabilité de nos résultats. Dans ce gel de données, nous avons également tenté d'améliorer la diversité des populations. L'étude de la génétique à travers des populations d'ascendance génétique diversifiée nous aide à mieux comprendre les variants génétiques affectant la gravité de la COVID-19 et leur impact à travers le monde. Sur les 47 études participantes, 19 incluaient des populations non européennes.
{kind=link}
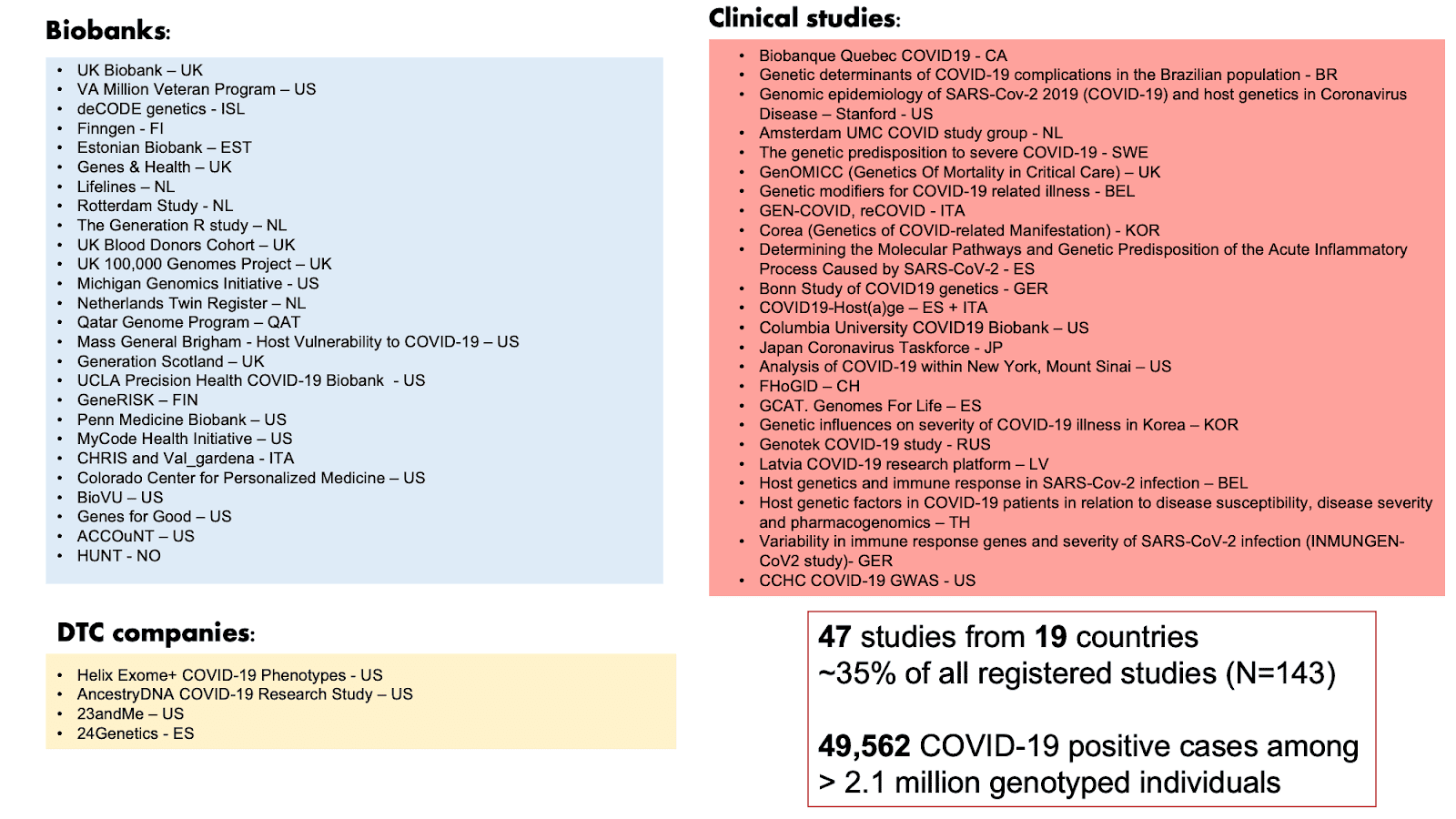
Figure 1: Liste des études participantes à la COVID-19 HGI pour la publication du gel de données 5. Sur les 47 études participantes, 19 incluaient des populations non européennes. Adapté de la présentation d'Andrea Ganna le 25 janvier 2021.
Structure de l’étude
Comme lors des gels de données précédents, nous avons poursuivi l’examen de trois issues (Figure 2): A) Clinique sévère liée à la COVID-19 (ventilation assistée ou décès), B) Hospitalisation pour COVID-19, C) Infection au SARS-CoV-2. Ces analyses visent à capturer les caractéristiques génétiques associées avec la susceptibilité au SARS-CoV-2 et la sévérité de la COVID-19. La dernière analyse (Analyse C) a pour but de détecter des variants génétiques contribuant à l’infection par le SARS-CoV-2. Elle inclut tous les cas, indépendamment de la présence de symptômes. Les résultats des analyses, les définitions des cas et des contrôles, et les tailles des échantillons sont décrits dans la Figure 2.

Figure 2: Définition des cas et des contrôles pour chaque analyse du gel de données 5. Notez que le SARS-CoV-2 est le virus qui cause la maladie infectieuse nommée COVID-19. Adapté de la présentation d’Andrea Ganna le 25 Janvier 2021.
Les régions du génome associées avec la COVID-19 pointent vers l’immunité innée et la dysfonction pulmonaire
Après la collecte des données génétiques produites par nos contributeurs, nous avons réalisé des GWAS selon les définitions de la Figure 2. Précédemment, lors du gel de données 4, nous avions mis en évidence de nouveaux signaux génétiques pour la susceptibilité et la sévérité de la COVID-19 dans 7 régions chromosomiques. Ces régions indiquent un possible rôle étiologique de l’immunité innée et de la dysfonction pulmonaire, ce qui est cohérent avec la compréhension clinique actuelle de la COVID-19. Lors du gel de données 5, nous avons identifié 15 régions génomiques significatives distribuées dans les chromosomes: 1 région chromosomique est significative à l’échelle du génome uniquement pour l’analyse centrée sur les patients en état critique (analyse A); 11 régions chromosomiques ont un effet plus important sur la sévérité que sur l’infection (analyse B); et 4 de ces régions sont spécifiques de la susceptibilité à être infecté par le virus (analyse C). Dans la Figure 3, nous présentons une représentation graphique de ces résultats sous forme de graphe de Miami (une version en panneaux du graphe de Manhattan. Appelé Miami car l’horizon des bâtiments de Miami se reflète dans l’eau).
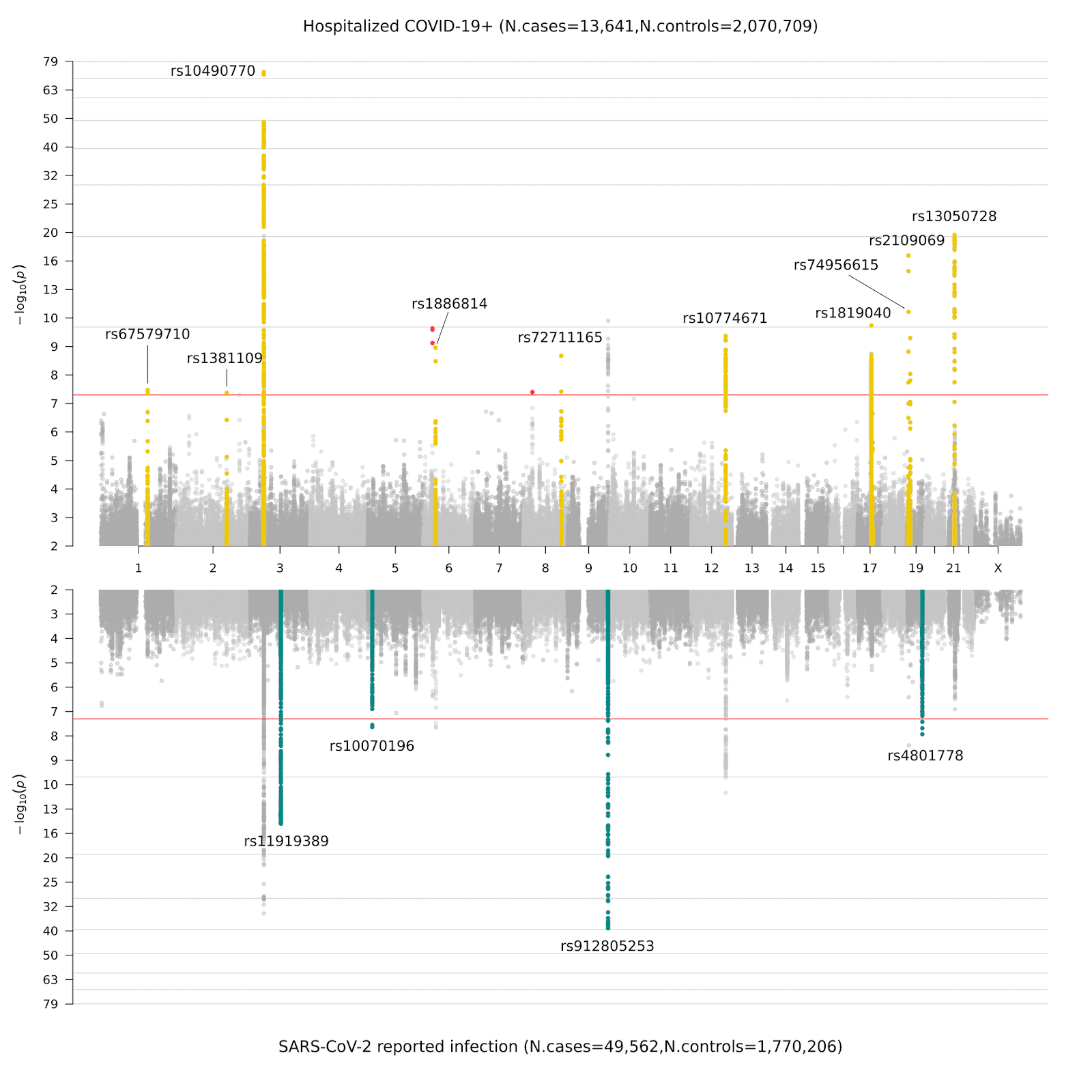
Figure 3. Graphe de Miami présentant les résultats des études d’association pangénomique pour la COVID-19. Le panneau supérieur montre les résultats de GWAS des patients hospitalisés pour COVID-19 versus contrôles (analyse B), le panneau inférieur les résultats de la GWAS pour infection au SARS-CoV-2 versus contrôles (analyse C).
Le rôle de la diversité des échantillons
Nous comprenons qu'avec de nombreuses études génétiques, la diversité dans la collecte d'échantillons est une préoccupation majeure (élaborée ici, en anglais). Comme tel, nous visions à améliorer la diversité de nos échantillons à mesure que notre étude se développait (Figure 4). L’amélioration de notre effort de collecte d'échantillons nous a conduit à identifier de nouveaux facteurs génétiques associés à la COVID-19 (nos résultats précédents sont discutés dans les articles de blog des versions 3 et 4). Grâce à l'identification concomitante de facteurs de risque génétiques à l'aide de nos méthodes d'analyse, nous sommes en mesure d'observer des variants génétiques au sein même ou à proximité de gènes. Jusqu'à présent, la plupart des gènes identifiés indiquent que l’augmentation du risque passe par des mécanismes cellulaires, la régulation immunitaire et la fonction cardiaque. L'identification de ces facteurs de risque pourrait conduire à des traitements ciblant ces gènes en particulier.
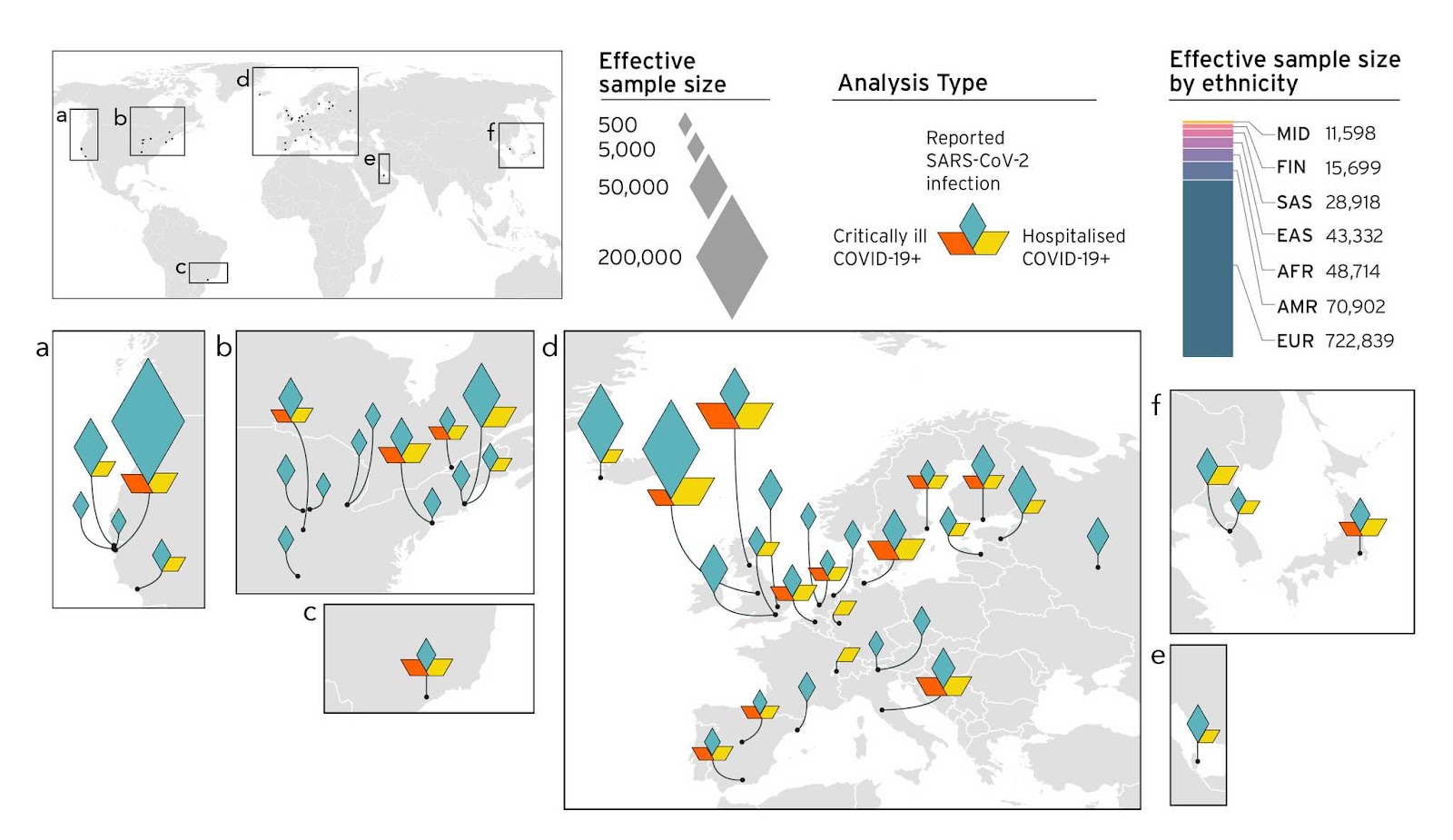
Figure 4. Aperçu des études contribuant à l'initiative de génétique de l'hôte de la COVID-19 et leur composition par principaux groupes d'ascendance dans les méta-analyses.
Dans le gel de données 5, 19 études ont contribué avec des populations non européennes: 7 incluant des afro-américains, 5 incluant des américains de multiple origines ancestrales, 4 incluant des asiatiques de l'est, 2 incluant des sud-asiatiques et une incluant des individus de descendance arabe. Les diamants montrent la taille effective de l'échantillon (taille de l'échantillon qui trouvera un effet statistiquement significatif) reçu de différents emplacements géographiques.
Nous avons trouvé 9 nouvelles régions chromosomiques associées à la COVID-19. Dans l'analyse A, pour laquelle nous cherchions des associations avec le développement de la maladie grave, il s'agit de régions chromosomiques proches de deux gènes: LZTFL1 sur le chromosome 3 et TAC4 sur le chromosome 17. La protéine LZTFL1 régule le trafic des protéines vers la membrane ciliaire (racine: cils). Les cils sont des structures ressemblant à des cheveux qui s'étendent du corps cellulaire vers l’espace intercellulaire. On les trouve dans les voies respiratoires, les poumons et de nombreux autres organes. LZTFL1 participe également aux réponses immunitaires. La protéine TAC4 a pour fonction de réguler la pression artérielle et le système immunitaire.
Pour l'analyse B, chez les patients hospitalisés avec la COVID-19, nous avons trouvé des associations avec des régions proches de 4 gènes. Tout d'abord, nous avons identifié une région chromosomique près de THBS3 sur le chromosome 1. Ce gène code pour la protéine THBS3 qui est exprimée dans le cœur et régulée à la hausse lors de maladies cardiaques. Deuxièmement, nous avons identifié une région chromosomique près de SCN1A sur le chromosome 2. Il a été démontré que des variations du gène SCN1A provoquent l'épilepsie et des convulsions. Troisièmement, nous avons identifié une région chromosomique près de TMEM65 sur le chromosome 8. Ce gène code pour la protéine TMEM65 qui joue un rôle dans le développement cardiaque, la régulation de la conduction et de la fonction cardiaque. Elle peut également jouer un rôle dans le métabolisme énergétique des cellules. Notamment, le variant identifié dans notre analyse pour TMEM65 a une fréquence allélique de 12% en Asie de l'Est et de 1% dans la population européenne. Les fréquences alléliques décrivent la variation observée à une position ou une région génomique dans une population donnée. Enfin, nous avons identifié une région chromosomique près de KANSL1 sur le chromosome 17. Il a été suggéré que la protéine codée par ce gène, KANSL1, joue un rôle dans les processus neuronaux.
Enfin, dans l'analyse C, qui cherche des associations avec les infections au SRAS-CoV-2 signalées, 3 nouvelles associations ont été trouvées dans les régions proches des gènes, ZBTB11 sur le chromosome 3, DNAH5 sur le chromosome 5 et PPP1R15A sur le chromosome 19. Premièrement, nous avons identifié une région par le gène ZBTB11 sur le chromosome 3. Ce gène code pour la protéine ZBTB11 dont il a été démontré qu'elle régule le développement des cellules immunitaires. Deuxièmement, nous avons identifié une région chromosomique dans ADNH5 sur le chromosome 5. Il a été démontré que les variations génétiques du gène ADNH5 provoquent dyskinésie ciliaire primaire, mouvement défectueux des cils entraînant des infections pulmonaires récurrentes, des symptômes aux oreilles/nez/gorge, bronchite et infertilité. Enfin, nous avons identifié une région chromosomique proche de PPP1R15A sur le chromosome 19. Ce gène code pour la protéine PPP1R15A qui est impliquée dans l'arrêt de la croissance et la mort cellulaire en réponse aux dommages à l'ADN, aux signaux de croissance négatifs et à une structure protéique incorrecte.
Dans nos analyses, les gènes qui affectent le système immunitaire jouent un rôle important dans la COVID-19. Les gènes impliqués dans la fonction pulmonaire et cardiaque et les processus neuronaux font également partie de nos découvertes. Des maladies cardiaques ont déjà été identifiées comme un facteur de susceptibilité à la COVID-19 et des symptômes neuronaux ont été rapportés dans le cadre de la COVID-19.
Corrélation ne signifie pas causalité
Les facteurs de risque identifiés dans les études d'association peuvent ne pas expliquer les causes de la susceptibilité à la COVID-19 ou sa gravité. Ainsi, nous avons utilisé une méthode appelée la Randomisation Mendélienne (RM) qui utilise des informations génomiques pour déterminer des associations causales. La RM est une méthode qui utilise des variants génétiques connus pour influencer une exposition donnée (par exemple, l'indice de masse corporel - IMC) pour examiner l'effet causal d'une exposition sur la maladie. Pour un examen plus approfondi, nous avons décrit la RM dans un article de blog récent (destiné au public scientifique). À travers les trois phénotypes de la COVID-19 évalués, nous avons identifié des associations causales statistiquement significatives entre ces trois phénotypes de la COVID-19 et 6 traits (sur les 38 traits testés, figure 4). Nous avons constaté qu'un indice de masse corporelle (IMC) plus élevé, prédit génétiquement, était associé à un risque plus élevé d'infection par le SRAS-CoV-2 et d'hospitalisation dû à la COVID-19. Ce résultat corrobore les résultats d'études observationnelles, qui ont rapporté que l’augmentation de l’IMC était associée à un risque accru de sévérité de la COVID-19.

Figure 5: Corrélations génétiques et estimations causales de randomisation mendélienne entre 38 traits et la sévérité du COVID-19 ou l’infection reportée au SARS-CoV-2. Les traits sont listés sur l’axe des X et les phénotypes de la COVID-19 sur l’axe des Y. Les corrélations génétiques (cg) négatives et les estimés causaux protecteurs issues de la randomisation mendélienne (RM) sont représentés en bleu, tandis que les corrélations génétiques positives et les estimés causaux associés au risque RM sont en rouge. Plus les carrés sont larges, plus la corrélation est significative. Les estimés causaux passant le seuil de significativité statistique sont symbolisés par un astérisque.
Un effort collaboratif mondial pour comprendre la génétique de l’hôte du COVID-19
Dans la crise mondiale actuelle de la pandémie COVID-19, ces résultats démontrent la puissance d'un effort mondial réunissant 47 contributeurs de tous horizons. Au total, nous avons identifié 15 régions génomiques associées à la susceptibilité et à la sévérité de la COVID-19. Pour étudier davantage la causalité de ces régions, nous avons utilisé l'inférence statistique (c'est-à-dire la randomisation mendélienne) pour identifier 8 traits avec une causalité statistiquement significative pour ces signaux GWAS de la COVID-19. Actuellement, nous finalisons nos résultats sous forme d’un article scientifique. Pendant que nous continuons à lutter contre la pandémie mondiale de COVID-19, la COVID-19 HGI continuera à produire des résultats génétiques de manière itérative. En travaillant ensemble de façon collaborative, nous pouvons identifier des signaux fiables nécessaires pour une meilleure compréhension des facteurs biologiques et de la présentation clinique de la COVID-19.
Remerciements
Nous souhaitons remercier Andrea Ganna, PhD pour ses retours et commentaires constructifs.